Advertise With UsAdvertise on our extensive network of industry websites and newsletters.
Advertise With UsAdvertise on our extensive network of industry websites and newsletters.
 Get the PBR newsletterSign up to our free email to get all the latest PBR
news.
Get the PBR newsletterSign up to our free email to get all the latest PBR
news.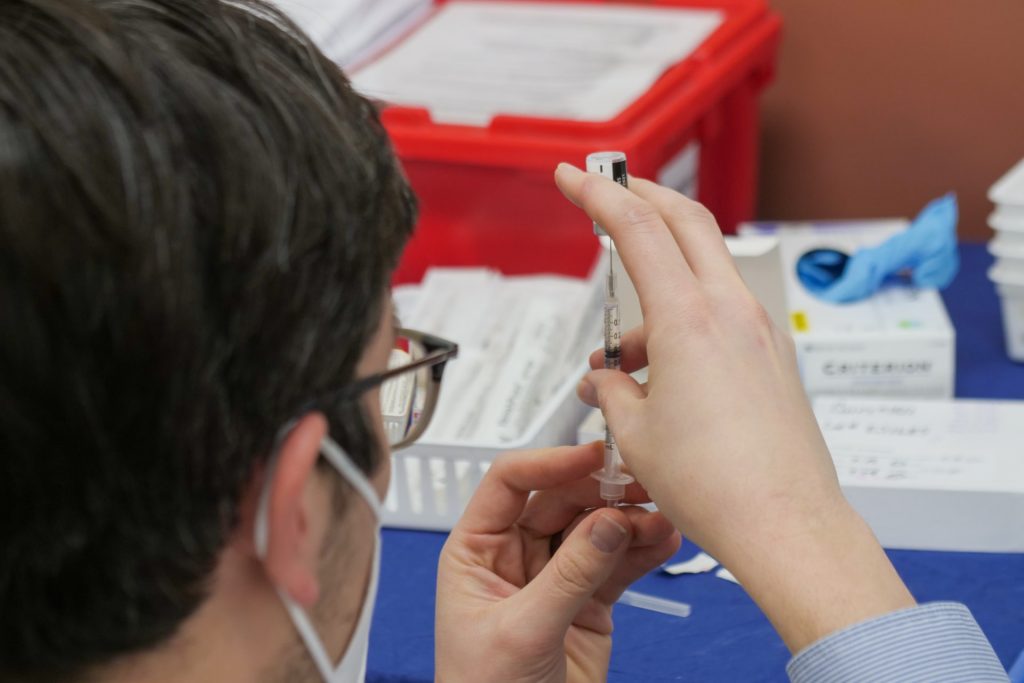
Johnson & Johnson to resume Covid-19 vaccine roll-out in Europe after EMA review
Johnson & Johnson (J&J) will resume Covid-19 vaccine roll-out in Europe following the European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC), which confirmed the overall benefit-risk profile being positive.